Case History:
A male patient in his mid-thirties with a personal and family history of polyps underwent duodenal polypectomy.
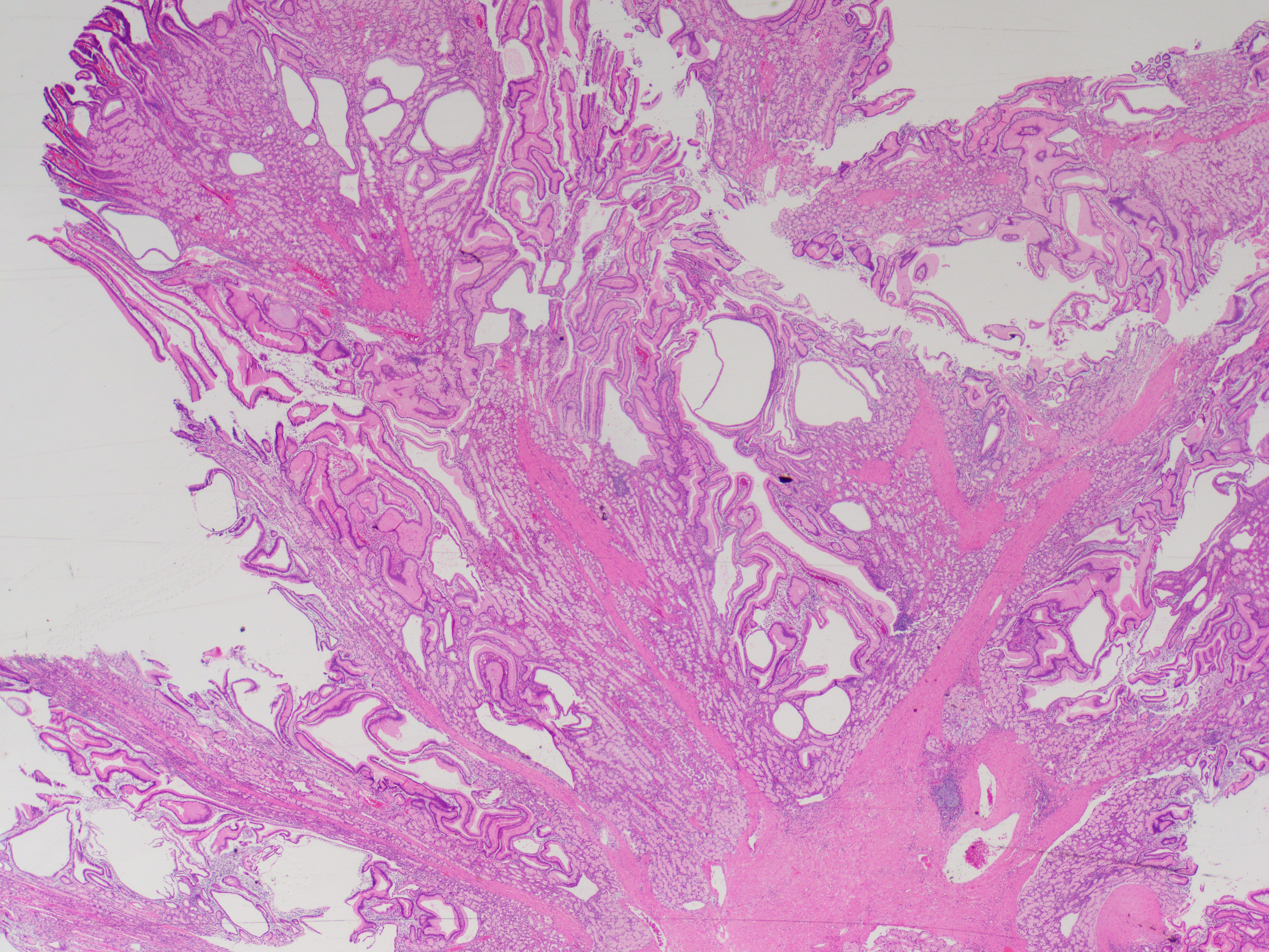
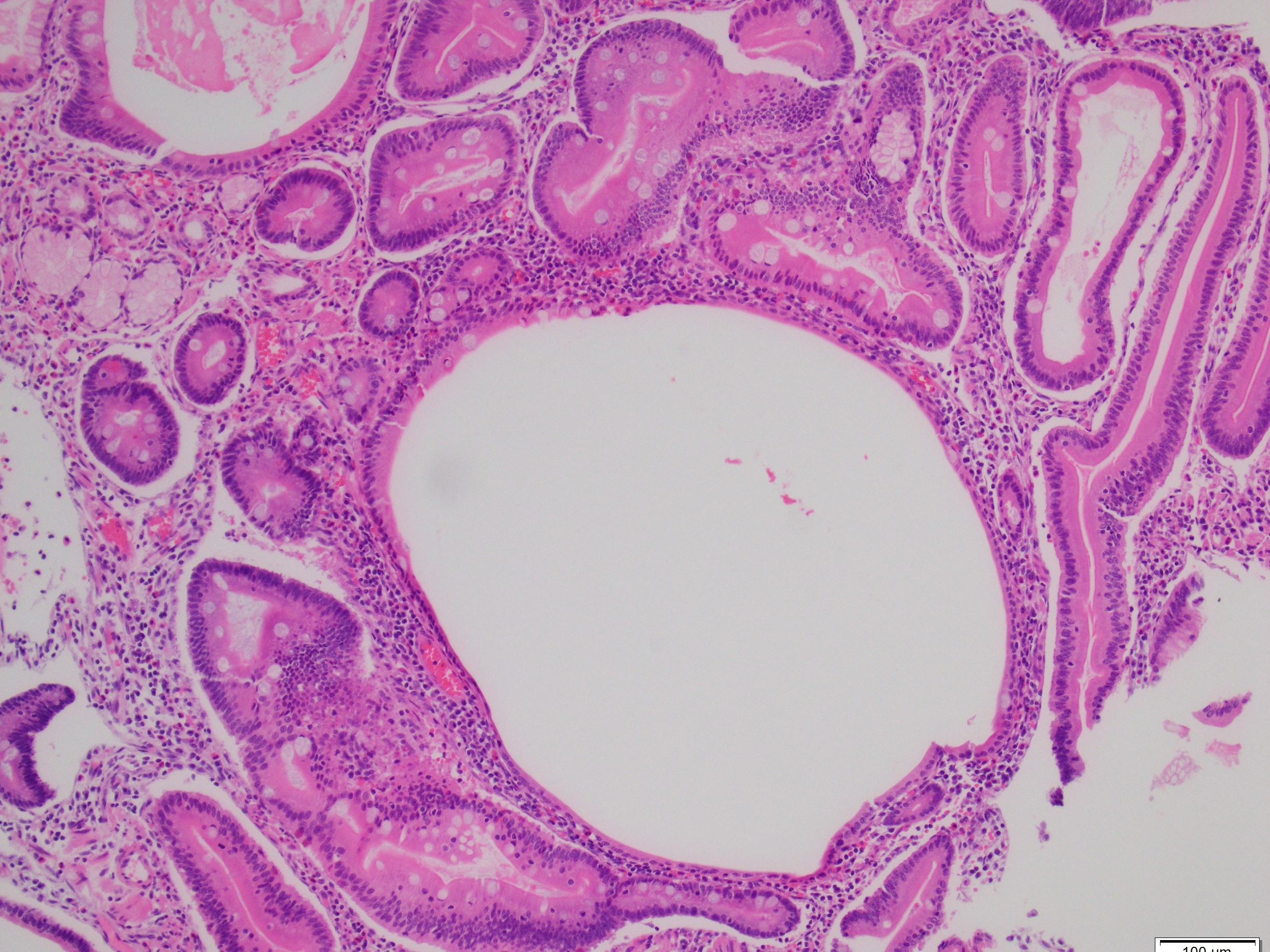
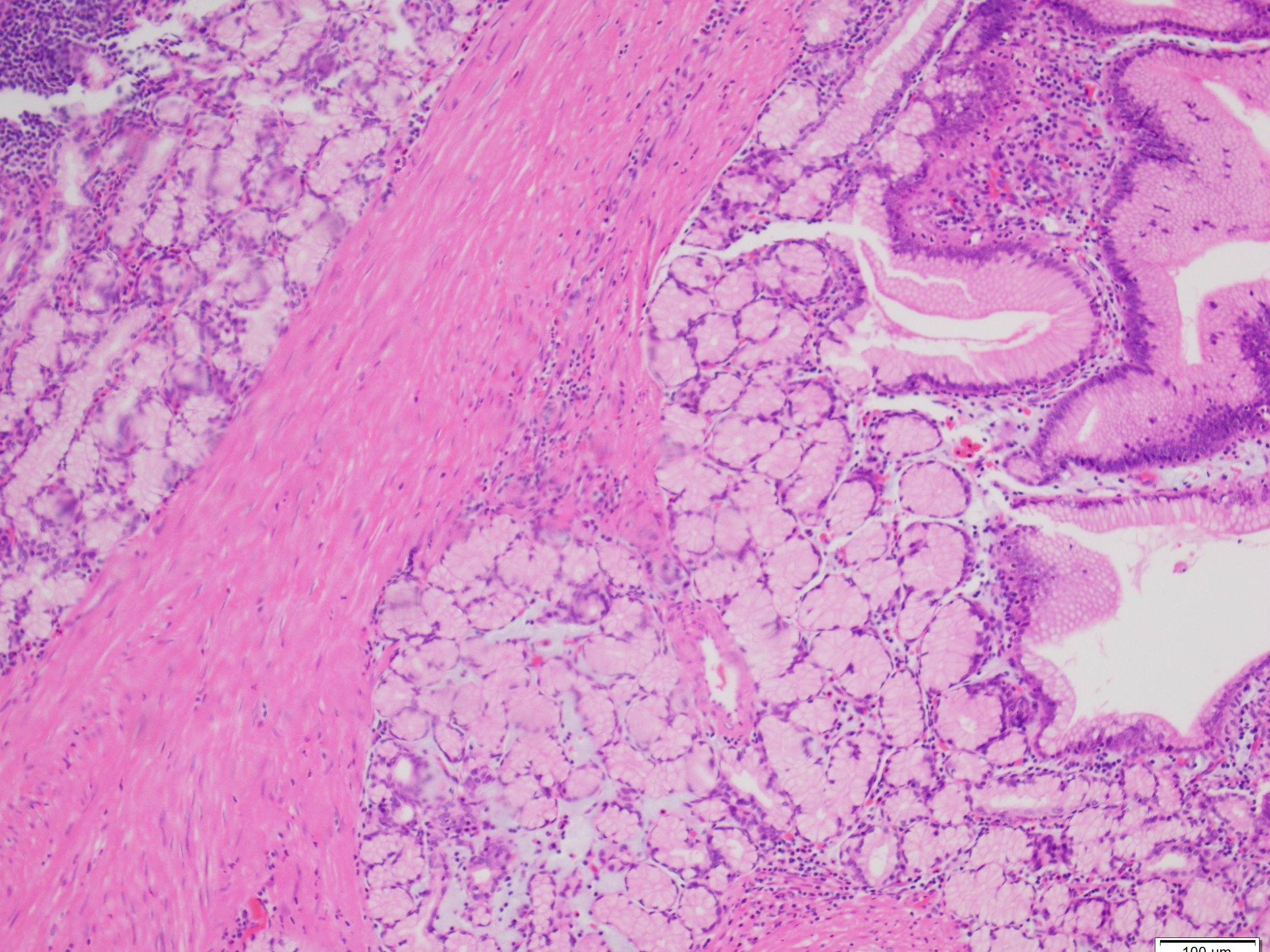
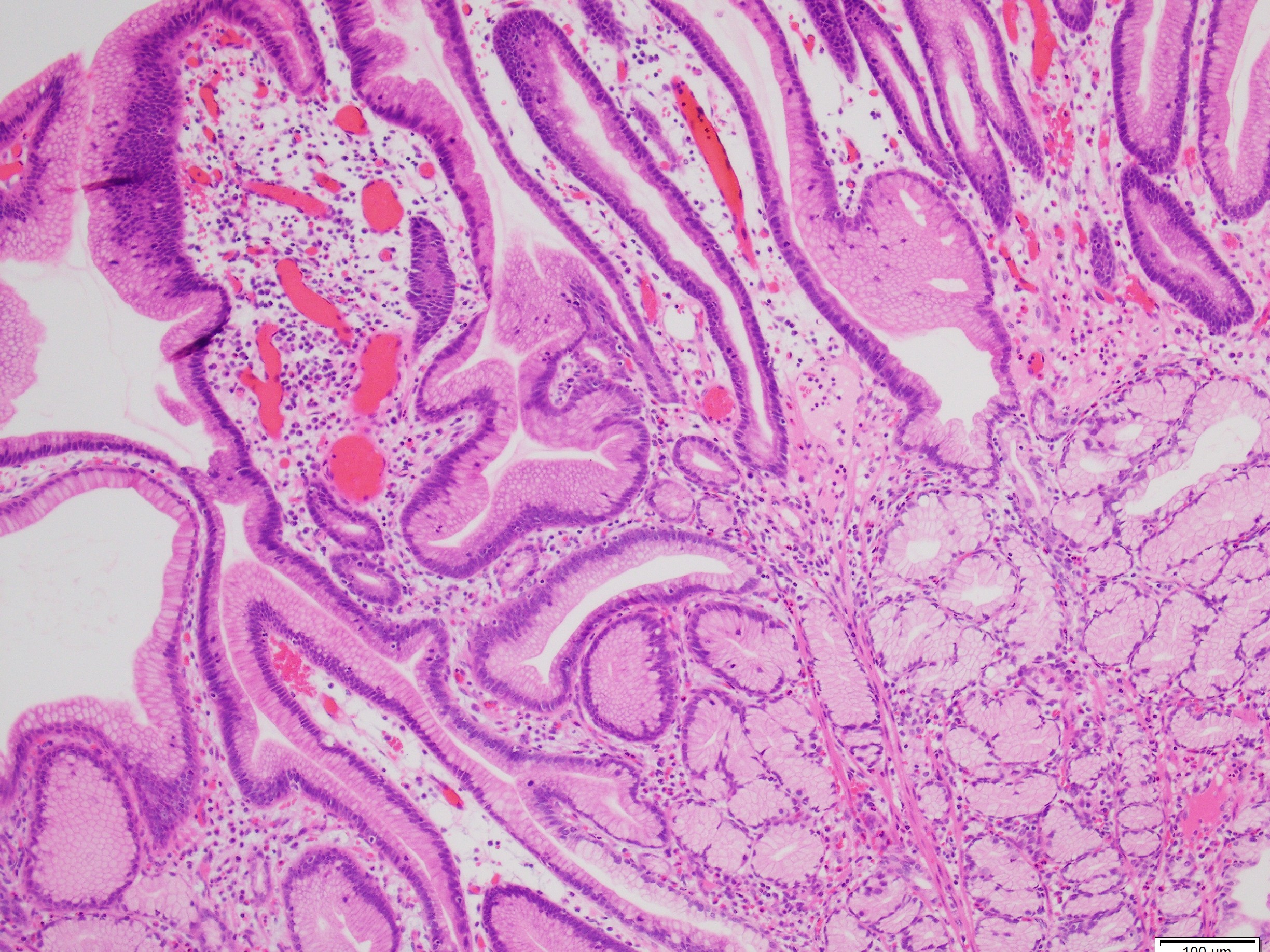
What is the diagnosis?
A. Peutz-Jeghers Syndrome
B. Juvenile Polyposis Syndrome
C. Cronkite-Canada Syndrome
D. Hereditary Mixed Polyposis Syndrome
Correct Answer: A. Peutz-Jeghers Syndrome
Discussion:
Peutz-Jeghers syndrome (PJS) is a rare, autosomal dominant hamartomatous polyposis syndrome characterized by the presence of distinctive gastrointestinal polyps and mucocutaneous pigmentation. It is caused by germline pathogenic variants in the STK11 gene (also known as LKB1), a tumor suppressor gene located on chromosome 19p13.3 (1). The most visible early sign of PJS is mucocutaneous pigmentation, which typically appears in infancy or early childhood. These pigmented macules—often dark brown or blue—are classically located on the lips, buccal mucosa, fingers, toes, and around the eyes or perianal region. While some of the skin lesions may fade with age, pigmentation in the mouth often persists into adulthood (1,2).
Histologically, PJS polyps are distinctive, showing an arborizing network of smooth muscle originating from the muscularis mucosae (Figures 1 and 3), with fronds of lamina propria and non-dysplastic epithelium (Figures 2-4)—features that can help distinguish them from other polyp types. However, their histologic overlap, particularly hamartomatous polyps and mucosal prolapse polyps, especially in the colon, can occasionally create diagnostic challenges (3,4). One of the most concerning aspects of PJS is its association with a substantially elevated lifetime risk for multiple malignancies. Cumulative cancer risk is estimated between 81%and 93% by age 65. Individuals with PJS are at increased risk for cancers not only of the gastrointestinal tract—such as colorectal, gastric, pancreatic, and small bowel—but also of the breast, ovary, cervix (adenoma malignum), and testes (Sertoli cell tumors). For example, the relative risk of pancreatic cancer may be over 100-fold higher than in the general population (1,2).
Juvenile Polyposis Syndrome (JPS): caused by SMAD4 or BMPR1A mutations, JPS features multiple juvenile polyps, mainly in the colon and stomach. Unlike PJS, it lacks mucocutaneous pigmentation and shows cystically dilated glands without arborizing smooth muscle. PTEN Hamartoma Tumor Syndrome (Cowden Syndrome): associated with PTEN mutations, this syndrome presents diverse GI polyps and extraintestinal features like macrocephaly and mucocutaneous papillomas but lacks PJS-specific pigmentation. Cronkite-Canada Syndrome: a non-hereditary syndrome with GI polyps, diarrhea, alopecia, and skin changes. Pigmentation is nonspecific and not confined to the lips or mucosa as in PJS. Mucosal Prolapse Polyps These reactive polyps may mimic the smooth muscle arborization of PJS but are typically solitary, left-sided, and occur in older patients. Hereditary Mixed Polyposis Syndrome (HMPS): features mixed polyp types due to GREM1 duplication. No pigmentation or typical PJS polyp architecture is present.
References:
1. Gammon A, Jasperson K, Kohlmann W, Burt RW. Hamartomatous polyposis syndromes. Best Pract Res Clin Gastroenterol. 2009;23(2):219-31.
2. Zbuk KM, Eng C. Hamartomatous polyposis syndromes. Nat Clin Pract Gastroenterol Hepatol. 2007 Sep;4(9):492-502.
3. Pai RK. When to suspect and how to diagnose syndromic polyps and carcinomas of the gastrointestinal tract: focus on Lynch syndrome and colonic hamartomatous polyposis. Diagn Histopathol 2019 Jan;26(1):8-14.
4. Rosty C. The Role of the Surgical Pathologist in the Diagnosis of Gastrointestinal Polyposis Syndromes. Adv Anat Pathol. 2018 Jan;25(1):1-13.
Case contributed by: Goo Lee, M.D., Ph.D., Associate Professor, Anatomic Pathology